
University of Nebraska-Lincoln
722 Hamilton Hall
Lincoln, NE 68588-0304
(402) 472-3039
rpowers3@unl.edu
Education
Postdoctoral, NIH-NIDDK, Bethesda, MD
Ph.D., Purdue University
B.A. Rutgers University
Research Interests
NMR spectroscopy, structural biology, metabolomics, bioinformatics, drug discovery
Current Research
Nuclear Magnetic Resonance (NMR) spectroscopy has proven itself an extremely versatile tool for exploring a variety of biological problems. NMR has been successfully used to determine the structure of numerous biomolecules, investigate biomolecular interactions, screen for potential drugs, determine structures of drug-complexes, monitor in vivo drug activity and analyze blood and urine for drug toxicity. As a result, NMR is widely used in the pharmaceutical industry to aid in drug discovery efforts. My research interest is focused on the application and development of NMR methodologies to understand the structure, function and evolution of novel proteins and their corresponding therapeutic utility in structure-based drug design programs.
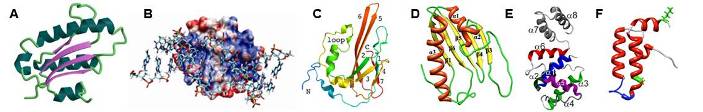
Examples of some of our recent NMR protein structures. (A) Archaeglobus fulgidis peptidyl-tRNA hydrolase, (B) Pseudomonas putida protein PpPutA45 and its DNA complex, (C) Pseudomonas aeruginosa protein PA1324, (D) protein YndB from Bacillus subtilis, (E) Staphylococcus aureus C-terminal domain of primase, and (F) J-domain of human DNAJA1.
Based on the recent success of the Human Genome project, the Structural Genomics initiative has established an ambitious goal of determining the structure of all the proteins in the genome. Approximately 30,000-90,000 proteins are predicted to be encoded from the human genome alone, where sequence similarity techniques may provide functional information for, at most, half of these proteins. To accomplish this goal will require the development of new techniques and methodology to rapidly determine protein structures and evaluate protein function. By combining the strengths of NMR, mass spectrometry and molecular modeling, rapid approaches to solving both protein and protein-ligand complexes are being explored in my group. Additionally, by modifying NMR screening techniques utilized to identify drug candidates, we are exploring the ability to quickly ascertain initial functional information for "hypothetical" (unknown function) proteins emerging from the Structural Genomics effort. Toward this end, we have developed the FAST-NMR technology that incorporates structural biology, ligand affinity screens and bioinformatics. Thus, the development of bioinformatics software and second generation databases are also an integral component of my research effort. The CPASS software and database was developed to facilitate protein functional annotation through the sequence and structure comparison of ligand-binding sites. Our PROFESS database is a second generation database that combines information about protein structure, function, sequence and evolution from multiple resource. PROFESS provides advanced query tools that enable the researcher to ask scientific questions and identify relationships not apparent from the individual data. This effort will be done in collaboration with the Northeast Structural Genomics Consortium(NESG).
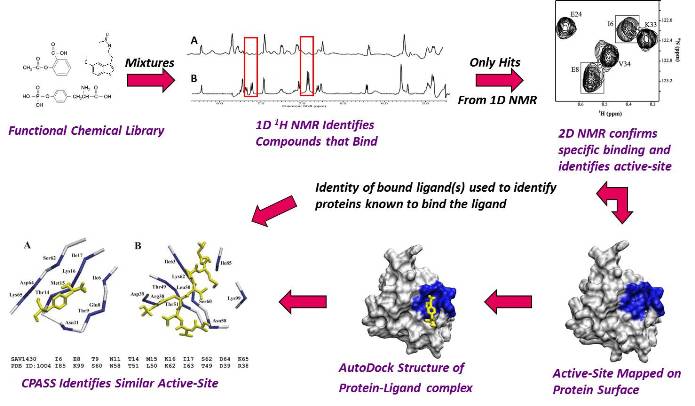
Flow Diagram of Our FAST-NMR Functional Annotation Methodology.
An emerging field for NMR has been its application in metabolomics, the analysis of metabolite changes. The approach is currently being used to identify disease markers and follow drug toxicity. Similar approaches can be used to identify protein function, follow in vivo drug activity or identify potential side-effects prior to clinical drug trials from the analysis of whole-cell extracts. We have developed a differential NMR metabolomics methodologyand the metabolomics phylogenetic-tree approach to monitor in vivo drug activity as part of our efforts towards the implementation of a systems biology approach to drug discovery. Our differential NMR metabolomics methodology is being broadly applied to explore various biological problems by applying systems biology. Again, these approaches will be beneficial in evaluating the function of hypothetical proteins associated with Structural Genomics.
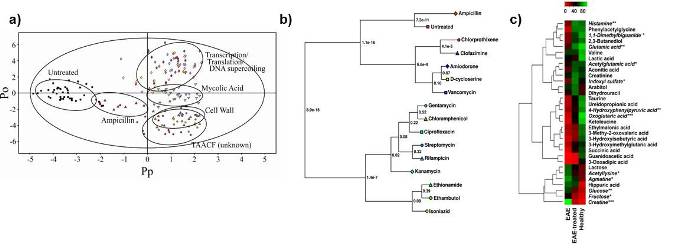
Examples of some of the typical approaches used in our lab to analyze NMR metabolomics data. (A) an OPLS-DA scores plot demonstrating the relative differences between classes. In this case, the different in vivo activity of TB drugs. (B) A metabolomics tree-diagram from our PCAtoTree software for the statistical analysis of clustering patterns in the scores plot from A. (C) Heatmap demonstring the relative changes in metabolite concentrations associated with Multiple Sclerosis.
For more information, please visit the Powers Research Group Homepage.
Selected Publications
(1)A. A. Crook, D. Zamora-Olivares, F. Bhinderwala, J. Woods, M. Winkler, S. Rivera, C. E. Shannon, H. R. Wagner, D. L. Zhuang, J. E. Lynch, N. R. Berryhill, R. C. Runnebaum, E. Anslyn, and R. Powers,* (2021)"Combination of Two Analytical Techniques Improves Wine Characterization by Vineyard, Region, and Vintage", Food Chemistry, 354, 129531 https://doi.org/10.1016/j.foodchem.2021.129531.
(2) R. Moore, T. Helikar, B. L. Puniya, R. Powers, B. Guda, K. W Bayles, and D. Berkowitz (2021) "Integrative network analyses of transcriptomics data reveal potential drug targets for acute radiation syndrome", Scientific Reports,11, 5585 https://doi.org/10.1038/s41598-021-85044-5.
(3) R. Poudel, F. Bhinderwala, M. Morton, R. Powers*, and D. J. Rose* (2021) "Metabolic Profiling of Historical and Modern Wheat Cultivars using Proton Nuclear Magnetic Resonance Spectroscopy", Scientific Reports,11, 3080 https://doi.org/10.1038/s41598-021-82616-3.
(4) A. A. Crook and R. Powers* (2020) "Quantitative NMR-based Biomedical Metabolomics: Current Status and Applications", Molecules, 25(21), 5128; https://doi.org/10.3390/molecules25215128..
(5) J. L. Catlett, J. Catazaro, M. Cashman, S. Carr, R. Powers, M. B. Cohen, and N. B. Buan* (2020) "Metabolic Feedback Inhibition Influences Metabolite Secretion by the Human Gut Symbiont Bacteroides thetaiotaomicron", mSystems, 5(5) e00252-20; https://doi.org/10.1128/mSystems.00252-20..
(6) B. Zhang, R. Powers, and E. ODay* (2020) "Evaluation of Non-Uniform Sampling 2D 1H-13C HSQC Spectra for Semi-Quantitative Metabolomics", Metabolites,10(5), 203; https://doi.org/10.3390/metabo10050203 PMC7281502.